NG重磅丨豆科植物泛基因组解析作物多样化分子机制
豆科植物(Fabaceae或Leguminosae)作为物种数量第三大的被子植物家族,是为人类提供必需蛋白质及通过固氮作用维持土壤肥力的关键资源,对可持续农业和全球粮食安全具有不可替代的作用。尽管豆科物种的进化历程已被广泛研究,但驱动适应性进化及调控农艺性状改良的保守机制仍不明确。
近日,南京农业大学宋庆鑫教授团队在国际著名期刊《Nature Genetics》上,发表了题为“Pangenome analysis provides insights into legume evolution and breeding”的研究论文,作者完成了九种广泛消费的食用豆类的高质量基因组组装,泛基因组分析揭示了冷季与暖季豆科作物中特定基因集的扩张现象,凸显了基因诞生与复制在根瘤自我调节过程中的作用。本研究揭示了豆科作物多样化的分子机制,并为加速豆类育种提供了宝贵资源。

一、豆类基因组从头组装
为探究豆科物种遗传多样性及适应性进化机制,作者对全球广泛消费的九种主要食用豆类进行了基因组从头组装,包括普通菜豆(苏彩豆19-17)、鹰嘴豆(XJ01)、豌豆(苏豌豆08)、小扁豆(SX01)、蚕豆(P17-186)、木豆(SM01)、豇豆(苏江豆01)、绿豆(苏绿豆07)和扁豆(扁红01)。
项目共生成732.7 Gb的PacBio HiFi测序数据(覆盖深度20-50×),针对鹰嘴豆,额外产生42.8 Gb ONT超长测序 ,结合Hi-C测序数据(约80-140×),最终获得所有物种染色体级别基因组组装,大小介于463.3 Mb至13.0 Gb之间。比较基因组学分析显示新组装与现有参考基因组间存在高度同线性保守,且多数组装质量优于已发表版本。
转座元件占基因组51-92%,作者共预测26,180-55,519个基因模型,其中绝大多数含有99%以上BUSCO参考基因集。相比已发表注释,新组装额外识别727-27,995个基因,超过88%的基因包含Pfam蛋白结构域。通过泛基因组分析,结合栽培大豆、野生大豆和白羽扇豆基因组,将全部基因划分为35,389个基因家族。随着基因组数量增加,基因家族总数上升而核心基因集呈相反趋势。按分布频率将基因家族分为四类:存在于全部12个基因组的12,436个核心基因家族;存在于10-11个基因组的1,794个软核心基因簇;存在于2-9个基因组的14,223个外壳基因集;以及6,936个物种特异性云基因簇。核心基因家族占基因总数41-70%。
与非必需基因相比,核心基因功能更保守:外类群中间源基因更多、Ka/Ks值更低、蛋白结构域注释比例更高。非必需基因的基因体内转座元件更多、富集于异染色质区、DNA甲基化水平更高,但染色质可及性和活性组蛋白标记(H3K4me3与H3K27ac)水平更低,表达量显著降低。核心基因主要富集于氮化合物生物合成、RNA生物合成等关键生物学过程,而非必需基因则富集于应激反应等过程。

图1:豆科植物泛基因组分析
二、根瘤共生相关基因的演化
植物与共生菌之间的根瘤共生现象零散分布于豆目、蔷薇目、壳斗目和葫芦目这四大植物类群,共同构成固氮分支植物(NFC)。关于根瘤共生的起源演化存在两种假说:(1)单次获得后大量丢失;(2)多物种独立演化形成。基于已发表RNA测序数据,作者在大豆中鉴定出1,305个根瘤优势表达基因,其中54%为核心基因。
根据系统发育树将大豆基因按物种间共享程度划分为0-7共8个等级,发现超过70%的根瘤优势表达基因存在于非NFC物种中。此外,81%以上已克隆的根瘤相关基因属于核心基因,其中90%以上在非NFC物种中存在。比较分析显示,固氮网络中的结瘤因子感知基因、钙信号传导基因以及根瘤特异性转录因子在被子植物、裸子植物、角苔和地钱中均保守存在,这些类群均能与丛枝菌根真菌形成共生。进一步发现55%的根瘤优势表达基因和77%的已克隆根瘤相关基因在裸子植物、角苔或地钱代表物种中保守存在。
通过豆科植物与11个外类群的比较分析,作者鉴定出358个豆科共有但外类群缺失的基因。经严格筛选,最终确定26个基因进行深入分析。其中8个基因可能起源于NFC演化阶段,14个起源于豆目阶段,4个起源于豆科阶段,其中5个基因在根瘤中高表达,包括大豆根瘤菌诱导的CLAVATA3/ESR相关肽编码基因GmRIC2。该基因通过负反馈机制(根瘤自动调节)抑制过量根瘤形成,表明固氮植物可能通过演化出新型系统反馈组件来维持碳氮供应的最优平衡。
系统发育分析将NIN基因划分为两个豆科特异分支:冷季豆类的NIN基因仅存在于分支1,而暖季豆类则具有2-4个拷贝且同时分布于两个分支。这种基因复制现象可能是暖季豆类对环境变化的适应性响应。

图2:豆科植物结瘤相关基因的演化
三、豆科进化过程中的基因扩张与丢失
冷季型与暖季型食用豆类的分化时间约在5500万年前。通过CAFE5软件分析,发现冷季豆类分支和暖季豆类分支从白羽扇豆分化后,分别出现40个和37个显著扩张的基因家族。另外仅有一个基因家族在两类豆科中共同扩张。尤其值得注意的是,冷适应关键基因CBF在冷季豆类中显著扩张。NLR蛋白作为植物抗病防御的核心组分,在暖季豆类中的丰度显著高于冷季豆类,这与高温环境下病原菌增殖及植物免疫抑制的生态特征相符。这些结果表明两类豆科采用不同的遗传策略适应各自生态位。
在古多倍体物种中,基因可分为WGD来源基因、小规模重复(SSD)基因和单拷贝基因。大豆属中鉴定出约31,900-33,700个WGD基因(Glycine-WGD基因),而其他暖季豆类仅保留2,800-3,500个豆科共同WGD基因(legume-WGD基因)。非必需基因在两类WGD基因集中占比均低于30%,但大豆谱系中约70%的单拷贝基因属于非必需基因,表明二倍化过程中非必需基因更易丢失其中一个拷贝。
在仅经历6500万年前WGD事件的暖季豆类中,SSD基因的软核心/外壳/云基因比例均高于WGD基因和单拷贝基因。鉴于非必需基因在应激响应中的关键作用,SSD基因中此类基因的比例提升可能补偿其优先丢失的缺陷,保障植物在胁迫环境中的生存优势。冷季豆类虽未经历近期WGD事件,但基因数量从鹰嘴豆的31,834个增至蚕豆的55,519个,这种增长主要源于SSD基因的扩张。通过比较分析发现,豌豆相对于鹰嘴豆新产生3,283个SSD基因,蚕豆相对于小扁豆新产生4,048个SSD基因,且近期形成的SSD基因中有显著更高比例源自古老WGD事件。

图3:豆科进化过程中的基因扩张与丢失
四、豆科进化中的趋同选择
保守基因的平行选择可导致不同物种获得相似表型,但豆科物种中与性状相关基因是否发生平行选择及其程度尚不明确。为此,作者利用大豆、木豆、鹰嘴豆和豌豆的野生种与地方品种/栽培种重测序数据,在独立演化约3000-5500万年的物种间鉴定平行选择基因组区域。
分析发现:大豆395个区域、木豆595个区域、鹰嘴豆470个区域及豌豆5,407个区域存在显著遗传多样性降低。其中226个大豆选择基因在木豆、鹰嘴豆和豌豆中均出现多样性降低,这些基因涉及种子休眠、籽粒膨大和能量平衡等通路。
基于SoyOmics数据库,我们利用3'UTR indel及下游SNP将GmYUC4a分为三种单倍型(Hap1-3)。携带Hap1的大豆材料籽粒重量显著低于Hap2/Hap3。等位基因频率分析显示:Hap1在野生大豆中占比94.5%,在地方品种中降至4.3%,在栽培种中进一步降至0.4%。对随机选取的10份携带Hap1或Hap2种质表达分析发现,Hap2携带者中GmYUC4a表达量更高。此外,在两个独立的染色体片段代换系R3和R170中,籽粒大小和重量均显著低于轮回亲本绥农14。这些结果表明YUC4基因可能在大豆、木豆、鹰嘴豆和豌豆中被趋同选择以增加种子重量。

图4:豆科进化中的趋同选择机制
五、转座元件的局部扩增驱动基因组扩张
TE扩增是基因组演化的重要驱动力。暖季豆类的TE扩增程度较轻,且主要集中于染色体中部区域;而冷季豆类则呈现大规模TE扩张。常染色质区域出现显著TE增殖,而异染色质区域总体TE比例保持稳定。
为解析基因组扩张中的TE扩增模式,作者在鹰嘴豆与最近缘物种豌豆间鉴定3,830个共线性区域,在小扁豆与最近缘物种蚕豆间鉴定3,185个共线性区域。采用"80-80-80"规则共识别到10个主要TE家族,其中F01家族在冷季豆类中拷贝数最高。F01与Ty3-gypsy Ogre元件序列相似度最高,是豌豆和蚕豆基因组中最丰富的TE类别。F01的扩张与基因间区延长呈正相关,鹰嘴豆中76.7%的无F01共线性区域在豌豆中被F01占据。而36%的小扁豆无F01区域在蚕豆中出现F01插入,但50%以上共线性区域在两物种中均含F01。这表明从鹰嘴豆到豌豆的基因组扩张主要由F01元件从头插入驱动,而从小扁豆到蚕豆的扩张则源于已有F01的扩增。
尽管TE广泛扩增,但其插入特异避开了基因体及侧翼0.25 kb区域。对此作者提出两种假说:(1)随机插入后通过自然选择清除基因区TE;(2)具有插入位点偏好的成簇整合。通过分析相邻F01间距发现:豌豆和蚕豆中分别有60%和70%的F01间距小于5 bp,显著短于随机插入模拟值,支持成簇插入假说。为进一步验证TE串联扩增,分析进化相关但基因组大小差异显著的二穗短柄草与小麦,小麦F01丰度更高,中位插入间距~410 bp,显著短于随机插入模拟值4,171 bp。这些结果表明基因稀疏区内TE的串联扩增可能是冷季豆类基因组扩张的重要机制,但自然选择清除基因区TE的可能性仍不能排除。
相较于鹰嘴豆,豌豆、小扁豆和蚕豆的染色体臂DNA甲基化水平显著升高,且呈均匀分布。F01元件呈现高CG甲基化水平,与其他沉默转座元件的模式相似。

图5:豆科进化中转座元件的扩张机制
六、调控元件的演化
TE具有演化为调控元件的潜力。通过ATAC-seq分析,在鹰嘴豆、豌豆、小扁豆和蚕豆中分别鉴定出22,507、63,137、56,696和98,458个开放染色质区域(OCRs)。作者将OCRs分为三类:与TE完全重叠的cTE-OCR、部分重叠的pTE-OCR以及无重叠的nTE-OCR(。cTE-OCR比例从鹰嘴豆的0.8%增至蚕豆的10%,且主要富集于远端调控区。而且豌豆、小扁豆和蚕豆的cTE-OCR呈现高DNA甲基化水平,但鹰嘴豆中未见此现象。
为探究OCR形成是否伴随DNA甲基化降低,作者将含OCR的TE分割为OCR区(开放)和非OCR区(关闭)。分析发现鹰嘴豆cTE-OCR区的DNA甲基化显著低于相邻非OCR区,但豌豆、小扁豆和蚕豆中两者甲基化水平相似,表明这些物种OCR形成不依赖DNA甲基化降低。
鹰嘴豆中92%以上OCR位于UMRs,而豌豆、小扁豆和蚕豆中仅不足半数OCR与UMRs重叠。此外,在三物种中分别鉴定出5,833、7,713和11,607个高CG甲基化的OCR。约28-45%的CG-OCR位于远端基因组区域,显著富集于DNA转座子。与随机基因组区域相比,远端CG-OCR具有显著更高的转录活性和染色质可及性。此外,发现7个染色质相关基因的表达量从鹰嘴豆到豌豆/小扁豆再到蚕豆逐步升高。这些高甲基化OCR的出现可能与豆科物种间染色质相关因子的差异表达有关。

图6:豆科调控元件的演化
结语
本研究通过对九种豆科物种进行基因组从头组装及泛基因组分析,系统解析了环境适应与人工选择背后的遗传变异基础。研究发现数百个基因在驯化过程中于多个豆科物种内同步出现遗传多样性降低,并提出转座元件"串联扩增"模型以解释冷季豆类的基因组扩张现象。这些发现为豆科进化机制提供了新见解,并为豆类遗传改良提供了宝贵的基因组资源。
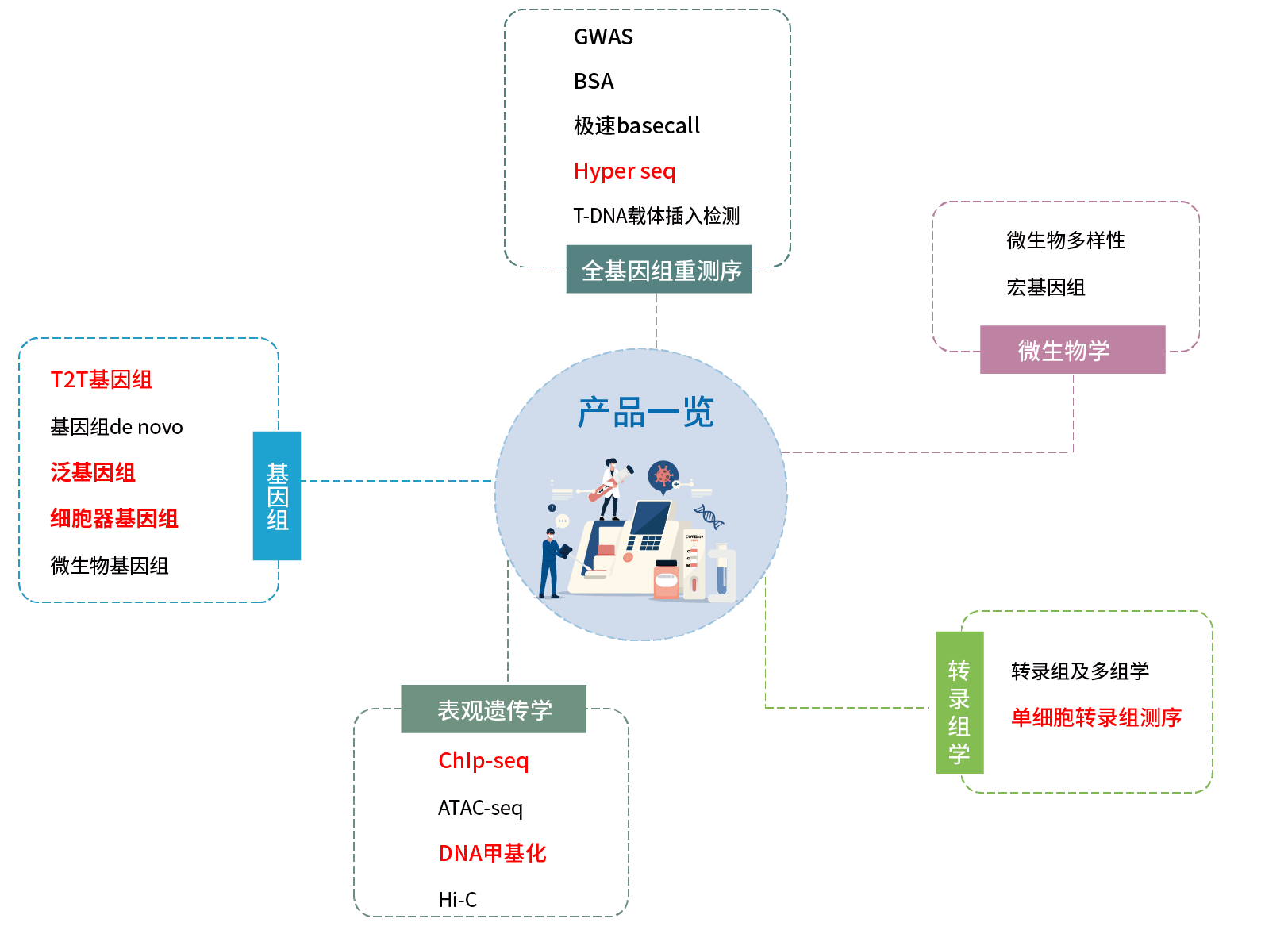
华命生物产品服务一览
华命生物目前已开通微信公众号、抖音、知乎、B站、小红书等线上平台,欢迎感兴趣的老师扫码关注了解更多内容!