Genome Biology | TRsv:准确检测TR-CNVs的高效新工具
基因组结构变异和短插入缺失是基因组中重要的变异类型,尤其是串联重复(TR)区域的拷贝数变异(TR-CNVs),它们通过重复单元的扩增或收缩形成多态性拷贝数等位基因。TRs包括短串联重复(STRs,微卫星)和可变数目串联重复(VNTRs),尽管只占基因组的3%,但由于高突变率,TR区域的插入(INS)和缺失(DEL)变异频繁发生,并与多种遗传病(如神经退行性疾病、癌症)相关。
国外研究团队于2025年8月20日在著名期刊Genome Biology中发表了一篇题为“TRsv: simultaneous detection of tandem repeat variations, structural variations, and short indels using long read sequencing data”的研究论文,报告了一个针对基因组中TR-CNVs检测的新软件,进一步基于138份Hifi数据绘制人群 TR-CNV 图谱,并揭示这些变异在基因调控、疾病和数量性状中的重要作用。

一、检测TR变异的问题
在TR变异检测中,常见的问题主要有两类,第一类出现在长读长序列比对到参考基因组时。由于串联重复区域本身高度重复,比对往往会被“切割”,在同一个区域内产生多个插入(INS)或缺失(DEL)的信号。这其实是一个TR变异,却被误判成多个小变异,导致结果碎片化、不准确。
第二类是非TR插入(non-TR-INSs)。即在TR区域里检测到的插入片段,并不是由TR单元组成的。它们分为两种情况:一是插入了与TR无关的序列,或只包含极少数重复单元;二是插入了移动元件(MEs),比如常见的 Alu、L1 和 SVA 等“跳跃基因”。研究发现,大约有 5% 的TR区域内插入属于这种非TR-INSs。

图1:串联重复区域(TR)中碎片化插入与缺失示例
二、TRsv的开发:检测TR-CNVs、SVs和Indels
作者开发TRsv来高效检测TR区域内外的TR-CNVs、SVs以及插入缺失。该工具利用基序/序列解析的方法,分析TR区域内的TR-CNVs,并识别TR区域内的ME插入、短SVs和插入缺失。通过结合来自UCSC和HipSTR站点的TR数据,TRsv在具有≥30%互惠重叠的TR区域中进行处理,限制大小为20bp到10Kb。TRsv能够识别TR-INS或DEL,并分配为不同单倍型的TR-CNVs。
对于非TR区域的INS,TRsv通过分析其与移动元件(MEs)的相似性、序列结构以及使用Tandem Repeat Finder(TRF)工具,识别含有串联重复的INS序列。低质量的SVs通过机器学习方法进行过滤。TRsv还具有合并来自多个样本的VCF文件功能,并能准确注释与基因区域重叠的SVs。

图2:TRsv算法概览
三、使用TRsv和其他现有工具评估TR - CNVs
作者系统评估了TRsv及现有工具在检测串联重复拷贝数变异(TR-CNV)中的性能,包括TR缺失(TR-DEL)、TR插入(TR-INS)和非TR插入(非TR-INS)。测试分为模拟数据和真实数据,在模拟数据评估中,研究人员基于GRCh38参考基因组构建了包含1000个人工TR-DEL、TR-INS和非TR-INS的数据集。结果显示,TRsv在三个变异类型上均达到接近100%的准确率和召回率,显著优于其他工具。LongTR虽在TR-DEL和TR-INS检测中表现良好,但对非TR-INS的准确率和召回率仅在20%左右。
在真实数据评估中,研究使用了GIAB HG002 TR-CNV目录的4746个TR位点数据集,包含3109个TR-INS、2416个TR-DEL和144个非TR-INS。TRsv再次展现出全面优势:TR-DEL(97.5%精度,94.4%召回)、TR-INS(97.4%精度,92.5%召回)、非TR-INS(93.9%精度,96.5%召回)。相比之下,其他工具如LongTR、TREAT和TRGT仅适用于特定变异类型检测。在运行效率方面,TRsv每CPU核检测1万个串联重复的速度位居前列,且能同时检测TR区域外的结构变异,在运行效率方面显著优于现有工具。
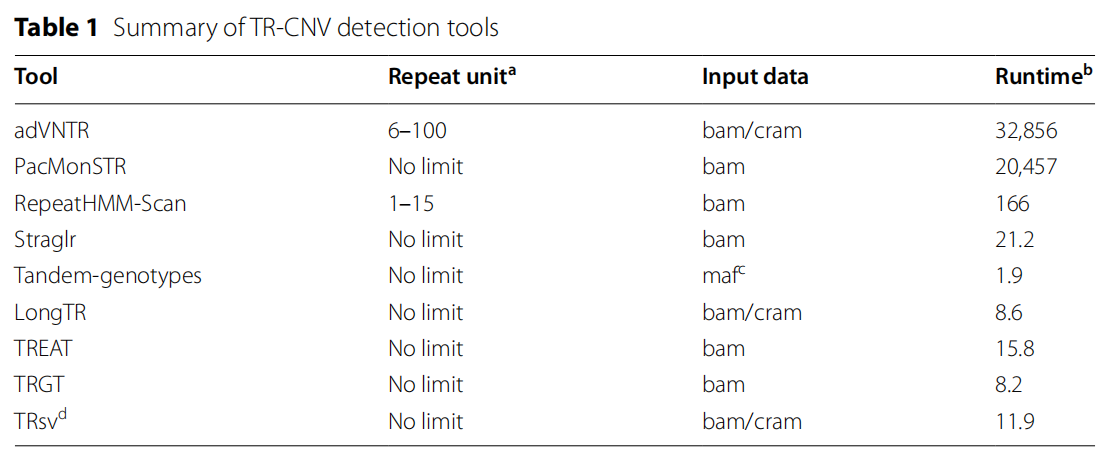
表1:TR-CNV检测工具汇总

图3:利用模拟数据和真实数据评估TR-CNV检测工具
四、TR区域外SV与短插缺变异的评估
作者基于NA12878 HiFi、HG002 HiFi和HG002 ONT三个数据集,进一步评估了TRsv与八种长读长SV检测工具在TR区域外的检测性能。结果显示TRsv在DEL检测中召回率最高(81.3-93.3%),INS检测表现(81.9-87.8%)与其他工具相当,且所有工具精度均达90-99%。人工验证表明TRsv假阳性极低,INS/DEL检测精度近100%。
TRsv通过机器学习过滤策略,显著提升了在非HiFi数据中的检测精度。在重复序列(DUP)检测方面,该工具展现出卓越性能:不仅对大于1Kb变异具有最高的真阳性检出率,同时保持了>96%的高基因分型精度。TRsv实现了1bp的超短插入及缺失检测,与PEPPER的一致性高达97%以上。随着变异片段增大,其召回率优势更为明显。这些突破性表现证实,TRsv在结构变异检测的灵敏度、精确度和变异覆盖范围等关键指标上均超越现有工具。
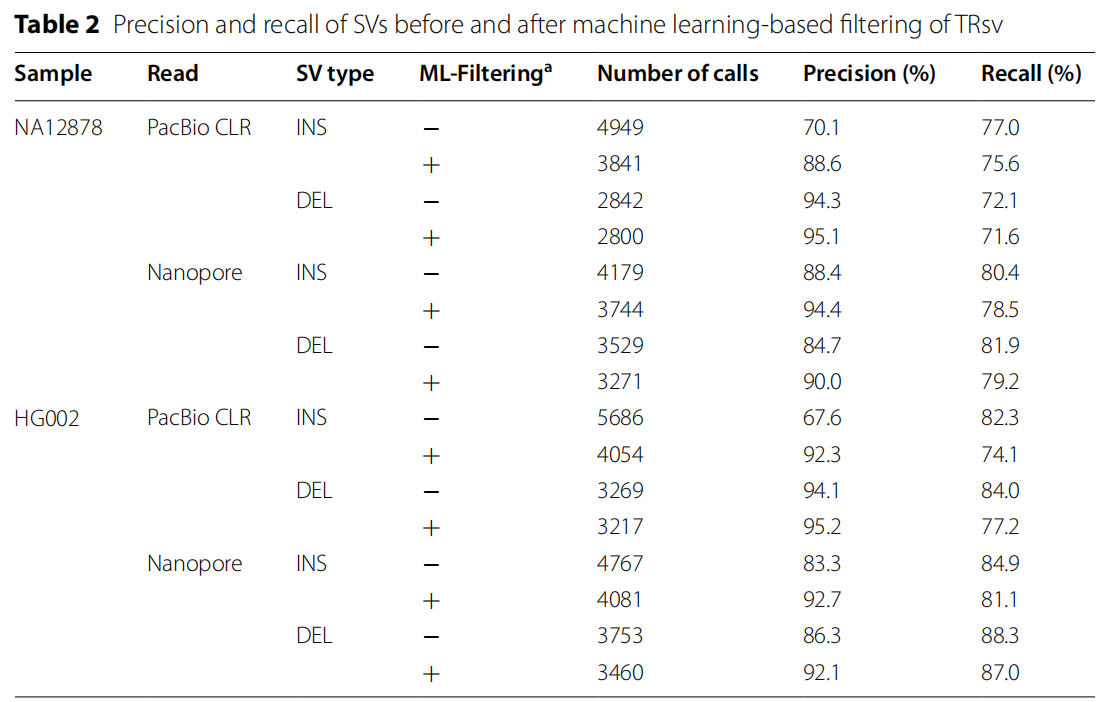
表2:在基于机器学习的 TRsv 过滤前后 SV 的精确率与召回率
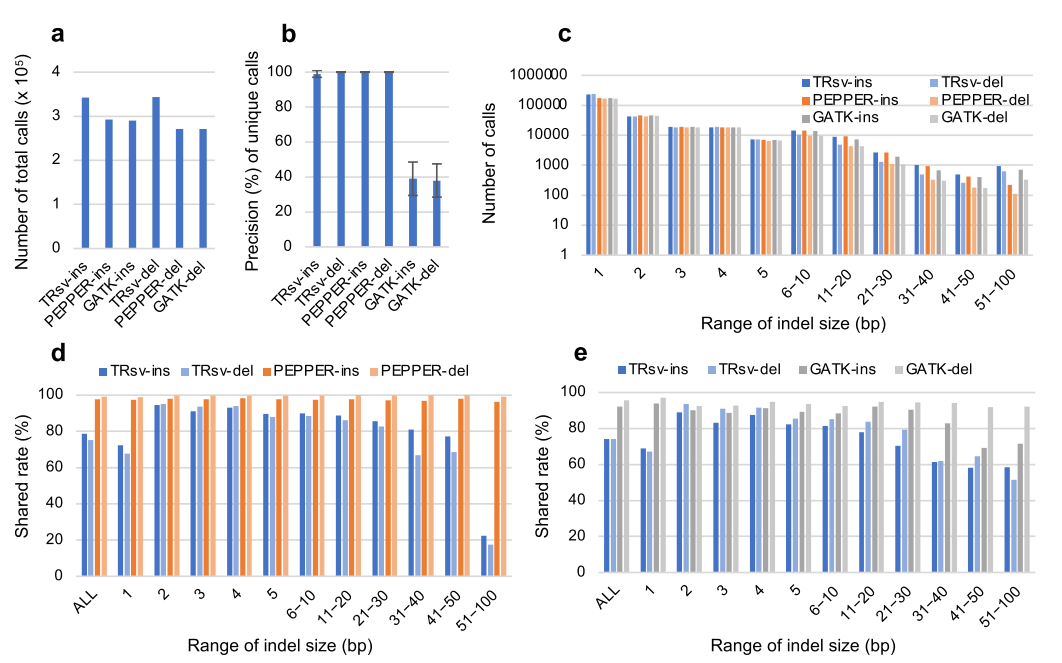
图4:TR区域外的短索引
五、基于138个PacBio HiFi数据集的TRsv变异检测
本研究利用138个Pac Bio HiFi全基因组测序数据集对TRsv工具进行了性能评估。结果显示,在严格的HiFi-var50标准下,共检测到60840个TR-CNV位点和78064个非TR区域的SV位点;在相对宽松的HiFi-var3标准下,检测结果大幅增加,分别为804415个TR-CNV位点和1599476个SV/indel位点。
进一步分析表明,在HiFi-var3条件下,约65%的TR-CNV位点表现为多等位性(即同一位点存在不同重复单位的变异);而在HiFi-var50条件下,这一比例下降至23%。此外,在39339个插入位点中,约一半(50.7%)为移动元件插入(ME-INS),其余则为包含不同重复单位的TR-INS。这些结果表明,TRsv在大规模基因组变异检测中表现出色,尤其在解析复杂的串联重复变异和不同类型的插入方面展现了强大优势。
六、三联体基序在重复扩增疾病中于CDSs和5′-UTRs富集
作者系统分析了不同长度重复序列(TR)及其拷贝数变异(TR-CNV)在基因区域中的分布特征及潜在功能。结果显示,在编码区(CDS)中,除3、6、9bp外的重复单元受到明显限制,反映了负向选择对移码翻译的抑制作用;而在5′UTR中,这类单元却显著富集,暗示其在翻译调控中可能发挥作用,并与多种疾病相关的重复扩增现象相呼应。在非编码基因外显子中也观察到一定富集,而内含子中则未呈现明显趋势。
进一步分析发现,GC富集的三联体基序(如CGG/GGC/GCG、CCG/CGC/GCC)在CDS与5′UTR中占据主导,并与疾病相关扩增高度一致;相比之下,内含子更常见的是AT富集的基序(如ATT、AAT)。此外,不同长度的TR-CNV在基序分布上也表现出差异:部分基序在大于50bp的TR-CNV中显著富集,而在较短片段中不明显。综上,三联体基序的拷贝数变异不仅取决于其自身的序列特性,还受到所在基因区域的影响。
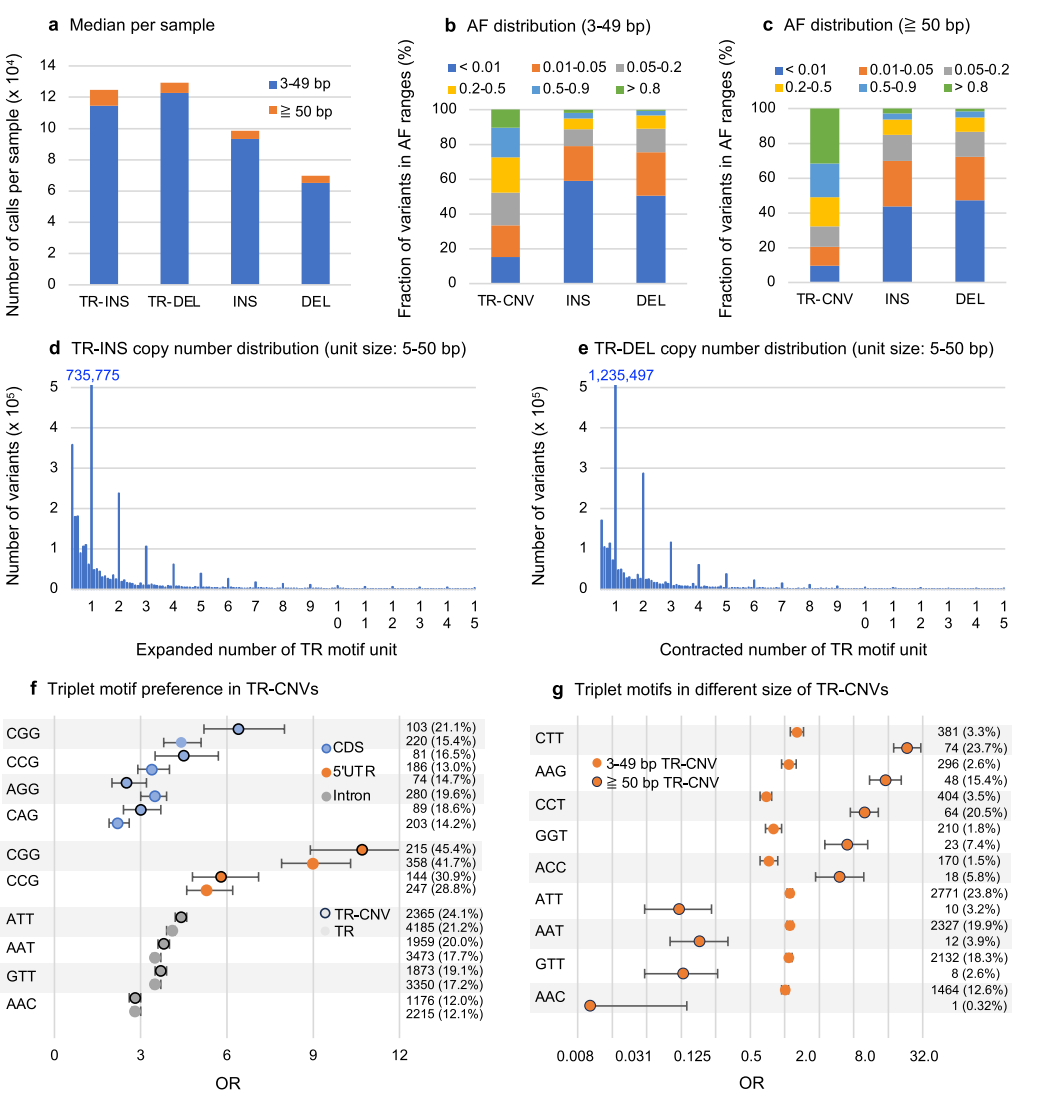
图5:从138个人类HiFi WGS数据中检测到的tr - cnv和SVs/ indes的特性
七、重复序列更常见于快速进化区和DNA修复热点
作者研究了100-5000 bp范围内的重复序列和拷贝数变异在功能基因组区域中的分布。结果显示,TR和TR-CNV显著富集于人类祖先快速进化区域(HAQERs)和双价染色质状态下的启动子区域(TssBiv)。其中,大于50 bp的TR-CNV在HAQERs中的富集更明显,且高等位基因频率(AF)的TR-CNV比低AF的更常见。与HAQER重叠的TR-CNV主要集中在500-1000 bp区间,符合HAQER的平均大小(约为800 bp)。相对而言,进化上保守的区域以及转录调控区域(如开放染色质、增强子和转录因子结合位点)对TR和TR-CNV表现出高度限制,说明这些功能区域大多缺乏TR。
进一步分析发现,TR和TR-CNV也富集于DNA复制和修复相关位点,如ATR激酶、MCM2/MCM7复制许可因子、ATM抑制诱导的断裂位点和复制起始点。这表明DNA复制压力可能导致复制叉在TR区域停滞,从而引发重复单元的扩增或收缩。然而,与同源重组(HR)修复相关的蛋白未见富集,提示这些损伤更可能通过非HR通路(如微同源介导末端连接,MMEJ)修复。
通过对538种蛋白的ChIP-seq分析,作者发现约15%的蛋白富集在TR-CNV区域,而在稳定TR中仅有4%。这些蛋白涉及DNA复制修复、染色质修饰、转录调控和RNA加工。这个结果表明,TR-CNV位点不仅是转录调控的热点,也是复制与转录压力下的DNA损伤易感区域。

图6:在138个HiFi WGS数据集中检测到功能基因组元件的TR-CNVs富集
八、与基因表达相关的TR-CNV在多种疾病和性状的基因区域中富集
研究最后通过整合59例人类样本的长读长测序和RNA-seq数据,系统解析了TR-CNV的调控功能。作者发现,大于20 bp的TR-CNV中存在104个显著影响基因表达的位点。深入研究表明,这些功能性TR-CNV主要富集在基因的外显子和启动子区域,并与转录活性区域高度重叠,特别是与RNA聚合酶复合物和RNA加工因子等关键调控元件共定位。
进一步分析揭示了这些TR-CNV在多种重要疾病相关基因中的显著富集现象,包括精神分裂症、帕金森病等神经系统疾病,以及多种癌症类型。同时,在血压、骨密度等复杂数量性状相关基因中也观察到类似的富集模式。这些发现为深入理解非编码变异在疾病发生和表型调控中的分子机制提供了新的研究视角。
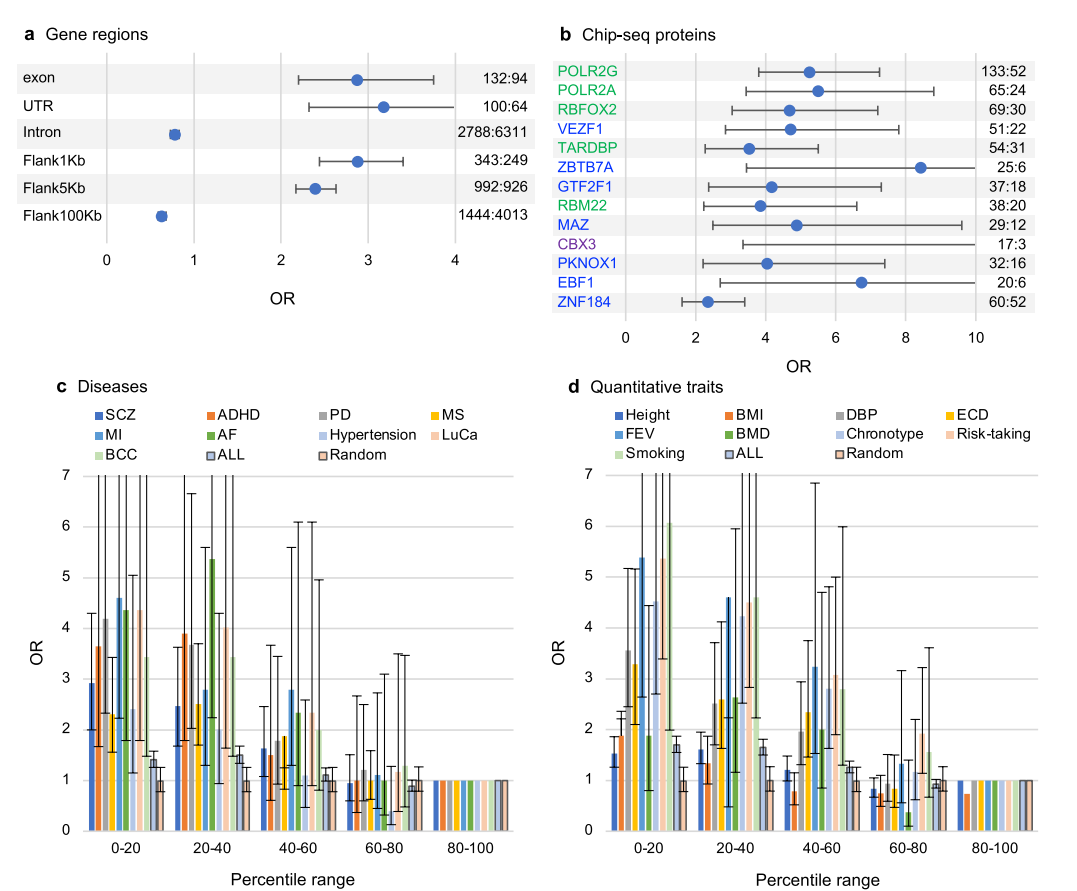
图7:与基因表达相关的eTR-CNVs
结语
本研究开发了一款新软件TRsv,可同时检测TR-CNV、SV以及indel。与同类工具相比,该软件在精度和召回率上均显著提升,且运行高效。基于138份HiFi数据,作者绘制了人群水平的TR-CNV图谱,发现个体普遍携带大量、多等位且等位频率较高的TR-CNV,并呈整数拷贝峰分布。功能分析显示,TR/TR-CNV显著富集于HAQER、复制起始点及DNA损伤应答位点,提示复制压力可能导致突变,并通过非HR通路修复。同时,GC富集的三联体重复倾向于分布在CDS和5′UTR区域。结合59份RNA-seq数据,研究进一步识别出与基因表达相关的eTR-CNV。这些变异在外显子和基因近端显著富集,能增强与转录及RNA加工因子的结合,并与多种常见疾病和数量性状相关。这些结果凸显了TR-CNV在基因调控与复杂性状形成中的重要性,也为相关疾病研究和潜在应用提供了新思路。
华命生物产品服务一览
华命生物目前已开通微信公众号、抖音、知乎、B站、小红书等线上平台,欢迎感兴趣的老师扫码关注了解更多内容!